A scalable approach to mapping out the blood cell landscape
Collaborative Melbourne research demonstrates that it is possible to combine a large number of different profiling experiments to create an integrated molecular map of human tissue.
Biological research technologies are evolving at a rapid rate and we are now able to peer inside cells with a high resolution. This information allows researchers to create nuanced processes to identify cell type and cell state through measurements such as expression, or activation, of certain genes. Just as photos are made up of pixels, researchers hope to collate data from many experiments to create a complete and complex map of different human tissue types.
However, combining data from many experiments is challenging, as each experiment is often performed with different technology or on different platforms, introducing artificial and unwanted effects on the data collation and analysis process.
Researchers from the University of Melbourne’s Centre for Stem Cell Systems and Melbourne Integrative Genomics demonstrated in A simple, scalable approach to building a cross-platform transcriptome atlas [LINK TO PAPER] published in JOURNAL, that it is possible to combine a large number of different profiling experiments to create an integrated molecular map of human tissue.
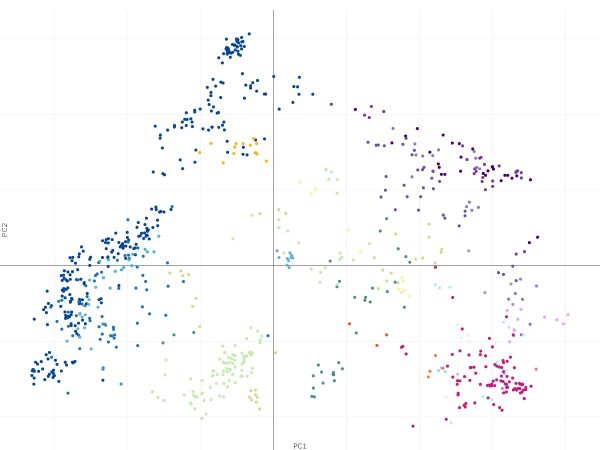
Led by Dr Paul Angel, a post-doctoral researcher in the Stem Cell Systems Lab, the paper introduces a simple and scalable method of data integration based on feature selection and the well-known principal component analysis (PCA) technique. These approaches allow for FAIR (findable, accessible, interoperable and reusable) data reuse and robust identification of molecular signatures across multiple studies, experimental conditions and platforms.
To test that the feature selection and PCA techniques were appropriate, Associate Professor Kim-Anh Lê Cao, Head of the Computational Statistics and Biology lab, and honours student Yidi Deng completed statistical analysis of the methods. They found that the feature selection technique was indeed selecting genes that were highly influenced by cell type, and not influenced by different technology.
This confirmed that there was a subset of reliably measured genes across different technological platforms and these genes could recapitulate the known relationships amongst blood cells, when the method was applied to blood data.
This blood atlas provides a reference for researchers who want to place their samples in the context of previous work by other labs. It also provides a reference point for new data types, such as the classification of single cells. The atlas is accessible for anyone to use at www.stemformatics.org/atlas/blood, and includes a host of easy-to-use tools and visualisations.
In the future, the team will work on expanding this methodology to create atlases for other cell and tissue types.